Title
A Phase 3 Randomized Study Comparing Daratumumab, Bortezomib, Lenalidomide and Dexamethasone (DVRd) followed by Ciltacabtagene Autoleucel versus Daratumumab, Bortezomib, Lenalidomide and Dexamethasone (DVRd) followed by Autologous Stem Cell Transplant (ASCT) in Participants with Newly Diagnosed Multiple Myeloma who are Transplant EligibleStudy map
Overview / Summary
This is a randomized, open-label, global, multicenter, Phase 3 study in adult participants with NDMM according to the International Myeloma Working Group (IMWG) diagnostic criteria for whom high-dose therapy and ASCT are part of the intended treatment plan.
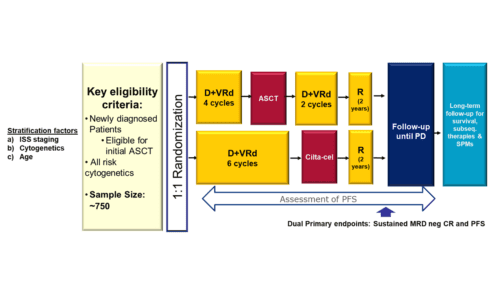
Participants randomized to Arm A will receive DVRd induction, followed by ASCT, DVRd consolidation, and lenalidomide maintenance therapy. Participants who discontinue study treatment for reasons other than documented disease progression (PD), death, or withdrawal of consent will enter the Post-treatment Follow‑up Phase during which they will continue to be monitored for efficacy, safety, and health-related quality of life (HRQoL) until confirmed PD, death, withdrawal of consent, lost-to-follow-up, or end of study, whichever occurs first.
Participants randomized to Arm B will receive DVRd induction, followed by cilta-cel infusion and lenalidomide post-CAR-T therapy. Participants will have intensive monitoring for efficacy, safety, pharmacokinetics (PK), biomarkers, and HRQoL during the first 112 days after cilta-cel infusion in the Post-infusion Follow-up Phase. Thereafter, participants will continue to be monitored until confirmed PD, death, withdrawal of consent, lost to-follow-up, or end of study, whichever occurs first in the Post-treatment Follow-up Phase.
Study details
Patient eligibility criteria
Approximately 750 participants (375 per arm) will be randomized in a 1:1 ratio into 2 arms.
Each potential subject must satisfy all of the following criteria to be enrolled in the study:
| 1. | 18-70 years of age at time of randomization, inclusive. | ||||||||||||||||
| 2. | Newly diagnosed participants for whom high-dose therapy and ASCT are part of the intended treatment plan with documented diagnosis of multiple myeloma according to IMWG diagnostic criteria (Rajkumar, 2014 Lancet Oncology). | ||||||||||||||||
| 3. | Measurable disease at Screening as defined by any of the following:
· Serum monoclonal paraprotein (M-protein) level ³1.0 g/dL or urine M-protein level ³200 mg/24 hours; or · Light chain multiple myeloma in whom the only measurable disease is by serum FLC levels in the serum: Serum immunoglobulin free light chain ³10 mg/dL and abnormal serum immunoglobulin kappa lambda free light chain ratio. Note: Local laboratory assessments may be used to establish measurable disease at Screening, with local laboratory result ³125% of requirements (eg, M-protein ³1.25 g/dL if using local labs). |
||||||||||||||||
| 4. | ECOG Performance Status grade of 0 or 1. | ||||||||||||||||
| 5. | Clinical laboratory values meeting the following criteria during the Screening Phase:
|
||||||||||||||||
| 6. | A woman of childbearing potential must have a negative highly sensitive serum pregnancy test (b‑human chorionic gonadotropin [b‑hCG]) at screening. | ||||||||||||||||
| 7. | When a woman is of childbearing potential, the following are required:
· Participant must agree to practice 2 methods of reliable birth control simultaneously from 4 weeks prior to initiating treatment with lenalidomide until 1 year after receiving a cilta-cel infusion or for 4 weeks following discontinuation of lenalidomide or for 3 months after discontinuation of daratumumab (whichever is later). One of the birth control methods should be a highly effective method of contraception (failure rate of <1% per year when used consistently and correctly; see examples below) and one other effective method (ie, male latex or synthetic condom, diaphragm, or cervical cap) and participant must agree to remain on both methods. Examples of highly effective contraceptives include: user-independent methods: 1) implantable progestogen-only hormone contraception associated with inhibition of ovulation; 2) intrauterine device; intrauterine hormone-releasing system; 3) vasectomized partner. user-dependent method: progestogen-only hormone contraception associated with inhibition of ovulation (oral or injectable). Estrogen-containing hormonal contraception is contraindicated due to increased risk of thromboembolic events with lenalidomide. women of childbearing potential must follow the contraception criteria outlined in the global REVLIMID® pregnancy prevention program or equivalent local REMS, whichever is more stringent, as applicable in their region. In addition to the highly effective method of contraception, a man: · Must always use a condom during any sexual contact with a woman of childbearing potential, even if they have undergone a successful vasectomy, from the time of signing the ICF until 1 year after receiving a cilta-cel infusion or for 4 weeks after discontinuing lenalidomide (whichever is later). · Who is sexually active with a woman who is pregnant must use a condom. · Should agree to practice contraception according to and for the time frame specified in the global REVLIMID® pregnancy prevention program or equivalent local REVLIMID® pregnancy prevention program, whichever is more stringent. Women and men must agree not to donate eggs (ova, oocytes) or sperm, respectively, during the study and for 1 year after receiving a cilta-cel infusion or for 4 weeks after discontinuing lenalidomide (whichever is later). Note: Hormonal contraception may be susceptible to interaction with the study treatment, which may reduce the efficacy of the contraceptive method. |
||||||||||||||||
| 8. | Participant must sign an ICF indicating that he or she understands the purpose of and procedures required for the study and is willing to participate in the study. Consent is to be obtained prior to the initiation of any study-related tests or procedures that are not part of standard-of-care for the participant’s disease. | ||||||||||||||||
| 9. | Willing and able to adhere to the prohibitions and restrictions specified in this protocol. | ||||||||||||||||