Title
A Phase 3 Study Comparing Daratumumab, VELCADE (bortezomib), Lenalidomide, and Dexamethasone (D-VRd) vs VELCADE, Lenalidomide, and Dexamethasone (VRd) in Subjects with Previously Untreated Multiple Myeloma who are Eligible for High-Dose Therapy. The Perseus StudyStudy map
Overview / Summary
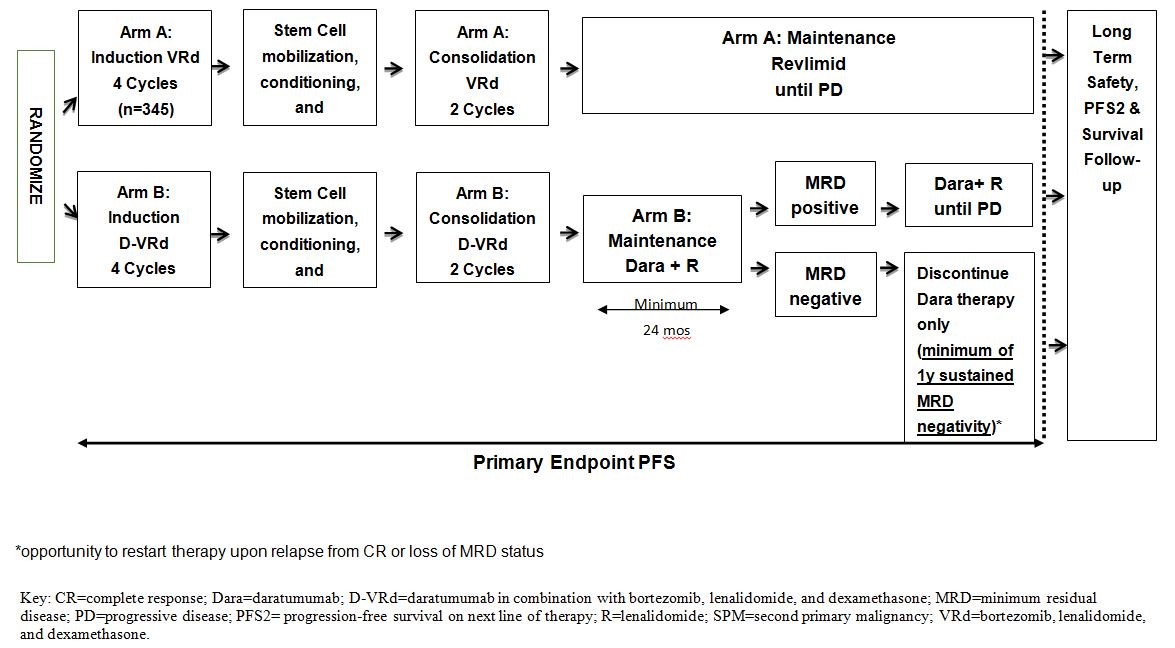
This is a randomized, open-label, multicenter study evaluating subjects with newly diagnosed multiple myeloma who are deemed eligible for high-dose therapy. Approximately 690 subjects will be stratified by International Staging System (ISS) Stage I, II, or III disease (β-2 microglobulin and albumin) and cytogenetics (standard risk or high risk as defined by presence of del17p, t[4;14] or t[14;16]), and then randomized in a 1:1 ratio. In Arm A, subjects will receive VRd for induction and consolidation, followed by lenalidomide (R) maintenance until disease progression. Subjects in Arm B will receive D-VRd for induction and consolidation followed by daratumumab and lenalidomide maintenance until disease progression. MRD-negative subjects in Arm B will stop therapy with daratumumab after sustained MRD negativity (at or below the threshold of 10-5) for 12 months and a minimum of 24 months of maintenance therapy. These subjects will continue lenalidomide maintenance therapy until disease progression. After stopping daratumumab therapy, subjects with sustained MRD negativity should restart therapy with daratumumab if there is a recurrence of MRD at 10-4 or higher or a confirmed loss of CR without disease progression, as evidenced by reappearance of serum or urine monoclonal protein (M-protein) or increase to ≥5% plasma cells in bone marrow After reinitiating daratumumab, the subject will continue daratumumab and lenalidomide therapy until disease progression.
Study details
Patient eligibility criteria
| Inclusion:
1.18 to 70 years of age, inclusive. 2.Monoclonal plasma cells in the bone marrow ≥10% or presence of a biopsy proven plasmacytoma and documented multiple myeloma satisfying at least one of the calcium, renal, anemia, bone (CRAB) criteria or biomarkers of malignancy criteria: CRAB criteria: 1. Hypercalcemia: serum calcium >0.25 mmol/L (>1 mg/dL) higher than upper limit of normal (ULN) or >2.75 mmol/L (>11 mg/dL) 2. Renal insufficiency: creatinine clearance <40mL/min or serum creatinine >177 μmol/L (>2 mg/dL) 3. Anemia: hemoglobin >2 g/dL below the lower limit of normal or hemoglobin <10 g/dL 4. Bone lesions: one or more osteolytic lesions on skeletal radiography, CT, or PET-CT Biomarkers of Malignancy: a. Clonal bone marrow plasma cell percentage ≥60% b. Involved: uninvolved serum FLC ratio ≥100 c. >1 focal lesion on magnetic resonance imaging (MRI) studies 3.Measurable disease as defined by any of the following: a. Serum monoclonal paraprotein (M-protein) level ≥1.0 g/dL or urine M-protein level ≥200 mg/24 hours; or b. Light chain multiple myeloma without measurable disease in the serum or the urine: Serum immunoglobulin FLC ≥10 mg/dL and abnormal serum immunoglobulin kappa lambda FLC ratio 4.Newly diagnosed subjects for whom high-dose therapy and autologous stem cell transplantation is part of the intended treatment plan. 5.Eastern Cooperative Oncology Group (ECOG) performance status score of 0, 1, or 2. 6.Clinical laboratory values meeting the following criteria during the Screening Phase (Screening hematology and chemistry tests should be repeated if done more than 3 days before C1D1): Adequate bone marrow function: a. Hemoglobin ≥7.5 g/dL (≥4.65 mmol/L; prior red blood cell [RBC] transfusion or recombinant human erythropoietin use is permitted however transfusions are not permitted within 7 days of randomization to achieve this minimum hemoglobin count); b. Absolute neutrophil count (ANC) ≥1.0 x 109/L (G-CSF use is permitted); c. Platelet count ≥50 x 109/L if bone marrow is >50% involved in myeloma. Otherwise ≥75 x 109/L Adequate liver function: a. Aspartate aminotransferase (AST) ≤2.5 x ULN; b. Alanine aminotransferase (ALT) ≤2.5 x ULN; c. Total bilirubin ≤1.5 x ULN (except in subjects with congenital bilirubinemia, such as Gilbert syndrome, direct bilirubin ≤1.5 x ULN) Adequate renal function: a. Estimated creatinine clearance ≥30 mL/min. Creatinine clearance may be calculated using Cockcroft-Gault, eGFR (Modified Diet in Renal Disease [MDRD]), or CKD-epi formula b. Corrected serum calcium ≤13.5 mg/dL (≤3.4 mmol/L); or free ionized calcium ≤6.5 mg/dL (≤1.6 mmol/L) 7. Female subjects of reproductive childbearing potential must commit to either abstain continuously from heterosexual sexual intercourse or to use 2 methods of reliable birth control simultaneously during the Treatment Period, during any dose interruptions, and for 3 months after the last dose of any component of the treatment regimen. Sexual abstinence is considered a highly effective method only if defined as refraining from heterosexual intercourse during the entire period of risk associated with the study drug. This birth control method must include one highly effective form of contraception (tubal ligation, intrauterine device [IUD], hormonal [birth control pills, injections, hormonal patches, vaginal rings or implants] or partner’s vasectomy) and one additional effective contraceptive method (male latex or synthetic condom, diaphragm, or cervical cap). Contraception must begin 4 weeks prior to dosing. Reliable contraception is indicated even where there has been a history of infertility, unless due to hysterectomy or bilateral oophorectomy. 8. A woman of childbearing potential must have 2 negative serum or urine pregnancy tests at Screening, first within 10 to 14 days prior to dosing and the second within 24 hours prior to dosing. 9. A woman must agree not to donate eggs (ova, oocytes) for the purposes of assisted reproduction during the study and for a period of 3 months after receiving the last dose of any component of the treatment regimen. 10. Male subjects of reproductive potential who are sexually active with females of reproductive potential must always use a latex or synthetic condom during the study and for 3 months after discontinuing study treatment (even after a successful vasectomy). 11. Male subjects of reproductive potential must not donate sperm during the study or for 3 months after the last dose of study treatment. 12. Signed an informed consent form (ICF) (or their legally acceptable representative must sign) indicating that he or she understands the purpose of, and procedures required for, the study and is willing to participate in the study. 13. Able to adhere to the prohibitions and restrictions specified in this protocol Exclusion: 1. Prior or current systemic therapy or SCT for any plasma cell dyscrasia, with the exception of emergency use of a short course (equivalent of dexamethasone 40 mg/day for a maximum 4 days) of corticosteroids before treatment. 2. Peripheral neuropathy or neuropathic pain Grade 2 or higher, as defined by the National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE) Version 5. 3. Prior or concurrent invasive malignancy (other than multiple myeloma) within 5 years of date of randomization (exceptions are adequately treated basal cell or squamous cell carcinoma of the skin, carcinoma in situ of the cervix or breast, or other non-invasive lesion that in the opinion of the investigator, with concurrence with the sponsor’s medical monitor, is considered cured with minimal risk of recurrence within 3 years). 4. Radiation therapy within 14 days of randomization. 5. Plasmapheresis within 28 days of randomization. 6. Clinical signs of meningeal involvement of multiple myeloma. 7. Chronic obstructive pulmonary disease (COPD) with a Forced Expiratory Volume in 1 second (FEV1) <50% of predicted normal (for subjects ≥65 years old FEV1 <50% or diffusing capacity of the lungs for carbon monoxide [DLCO] <50%) 8. Moderate or severe persistent asthma within the past 2 years, or currently has uncontrolled asthma of any classification. (Note that subjects who currently have controlled intermittent asthma or controlled mild persistent asthma are allowed in the study). 9. Any of the following: a. Seropositive for human immunodeficiency virus (HIV) b. Seropositive for hepatitis B (defined by a positive test for hepatitis B surface antigen [HBsAg]). Subjects with resolved infection (ie, subjects who are positive for antibodies to hepatitis B core antigen [antiHBc] and/or antibodies to hepatitis B surface antigen [antiHBs]) must be screened using real-time PCR measurement of hepatitis B virus (HBV) DNA levels. Those who are PCR positive will be excluded. EXCEPTION: Subjects with serologic findings suggestive of HBV vaccination (antiHBs positivity as the only serologic marker) AND a known history of prior HBV vaccination, do not need to be tested for HBV DNA by PCR. c. Seropositive for hepatitis C (anti-HCV antibody positive or HCV-RNA quantitation positive), except in the setting of a sustained virologic response (SVR), defined as aviremia at least 12 weeks after completion of antiviral therapy. 10. Concurrent medical or psychiatric condition or disease (such as but not limited to, systemic amyloidosis, POEMS, active systemic infection, uncontrolled diabetes, acute diffuse infiltrative pulmonary disease) that is likely to interfere with the study procedures or results, or that in the opinion of the investigator, would constitute a hazard for participating in this study. 11. Any of the following: a. myocardial infarction within 6 months before randomization, or an unstable or uncontrolled disease/condition related to or affecting cardiac function (eg, unstable angina, congestive heart failure, New York Heart Association Class III-IV) b. uncontrolled cardiac arrhythmia or clinically significant electrocardiogram (ECG) abnormalities c. screening 12-lead ECG showing a baseline QT interval >470 msec d. left ventricular ejection fraction (LVEF) <40% for subjects age 65-70 years old 12. Received a strong CYP3A4 inducer within 5 half-lives prior to randomization (Flockhart 2016: http://medicine.iupui.edu/flockhart/) 13. Allergy, hypersensitivity, or intolerance to boron or mannitol, corticosteroids, monoclonal antibodies or human proteins, or their excipients (refer to the Investigator’s Brochure), or sensitivity to mammalian-derived products or lenalidomide. 14. Not able to comply with the study protocol (eg, because of alcoholism, drug dependency, or psychological disorder). Subject has any condition for which, in the opinion of the investigator, participation would not be in the best interest of the subject (eg, compromise the well-being) or that could prevent, limit, or confound the protocol-specified assessments. 15. Pregnant, or breast-feeding, or planning to become pregnant while enrolled in this study or within 3 months after the last dose of any component of the treatment regimen. Or, subject is a man who plans to father a child while enrolled in this study or within 3 months after the last dose of any component of the treatment regimen. 16. Major surgery within 2 weeks before randomization or will not have fully recovered from surgery, or has surgery planned during the time the subject is expected to participate in the study. Kyphoplasty or Vertebroplasty is not considered major surgery. 17. Received an investigational drug (including investigational vaccines) or used an invasive investigational medical device within 4 weeks before randomization or is currently enrolled in an interventional investigational study. 18. Contraindications to the use of any components of the backbone treatment regimens, per local prescribing information. 19. Gastrointestinal disease that may significantly alter the absorption of oral drugs 20. Vaccination with live attenuated vaccines within 4 weeks of first study agent administration 21. Unable or unwilling to undergo antithrombotic prophylactic treatment. |